
AI 에이전트로 scRNA-seq QC, ‘스킬’ 쓰면 재현성이 달라질까? (Part 1)
요즘은 “데이터 던져주고 QC 해줘” 하면 AI 에이전트가 알아서 scanpy 코드를 짜고 돌려줍니다. 그런데 문득 궁금해졌습니다.
AI 에이전트한테 scRNA-seq 분석을 시킬 때, bioinformatics ‘스킬’을 쓸 때랑 그냥 맡길 때랑 질적으로 차이가 있을까? 특히 재현성(reproducibility) 과 정확성에서.
말로만 “스킬 쓰면 좋아요~” 하긴 싫어서, Claude Code로 직접 실험해봤습니다. 결론부터 말하면 — 재현성에서는 차이가 꽤 명확했고, 정확성에서는 생각보다 미묘했습니다. 숫자로 보여드릴게요.
💡 여기서 ‘스킬’은 Claude Code의
single-cell-rna-qc스킬입니다. scverse best practice를 따르는 MAD 기반 QC 파이프라인을 고정된 파이썬 스크립트(qc_analysis.py)로 제공합니다.
실험 설계
공정한 비교가 핵심이라, 조건을 최대한 깔끔하게 맞췄습니다.
- 데이터:
scanpy의 고전 데이터셋pbmc3k— 사람 PBMC, 2,700 cells × 32,738 genes (raw counts). 누구나sc.datasets.pbmc3k()로 받을 수 있어 재현 가능합니다. - 작업 범위: QC만 (load → QC metric 계산 → 저품질 셀/유전자 필터링 → 저장). 스킬이 다루는 범위와 정확히 일치시켰습니다.
- 두 조건:
| 조건 | 실행 방식 | 측정 |
|---|---|---|
| 스킬 미사용 | 독립 서브에이전트 5회 — 동일한 중립 프롬프트(“best practice로 QC 해줘”), 방법론·임계값 일절 미지정, 각자 격리된 디렉토리 | 5회 결과의 분산 |
| 스킬 사용 | 결정적 스크립트 qc_analysis.py를 2회 실행 |
분산 = 0 대조군 |
스킬 미사용 에이전트들에게는 스킬의 존재도, 이 실험의 의도도 알려주지 않았습니다. “QC 어떻게 할지는 네 판단대로.” 딱 이 정도. 진짜 자연스러운 변이를 보고 싶었거든요.
⚖️ 왜 스킬은 2회만? 스킬은 고정된 스크립트라 같은 입력이면 같은 출력이 나올 수밖에 없습니다. 그래서 “정말 byte 단위로 같은가”만 확인하면 충분합니다. 반대로 미사용 조건은 매번 새로 코드를 짜므로 분산을 보려면 여러 번 돌려야 하고요.
결과 1 — 재현성: 여기서 갈렸습니다
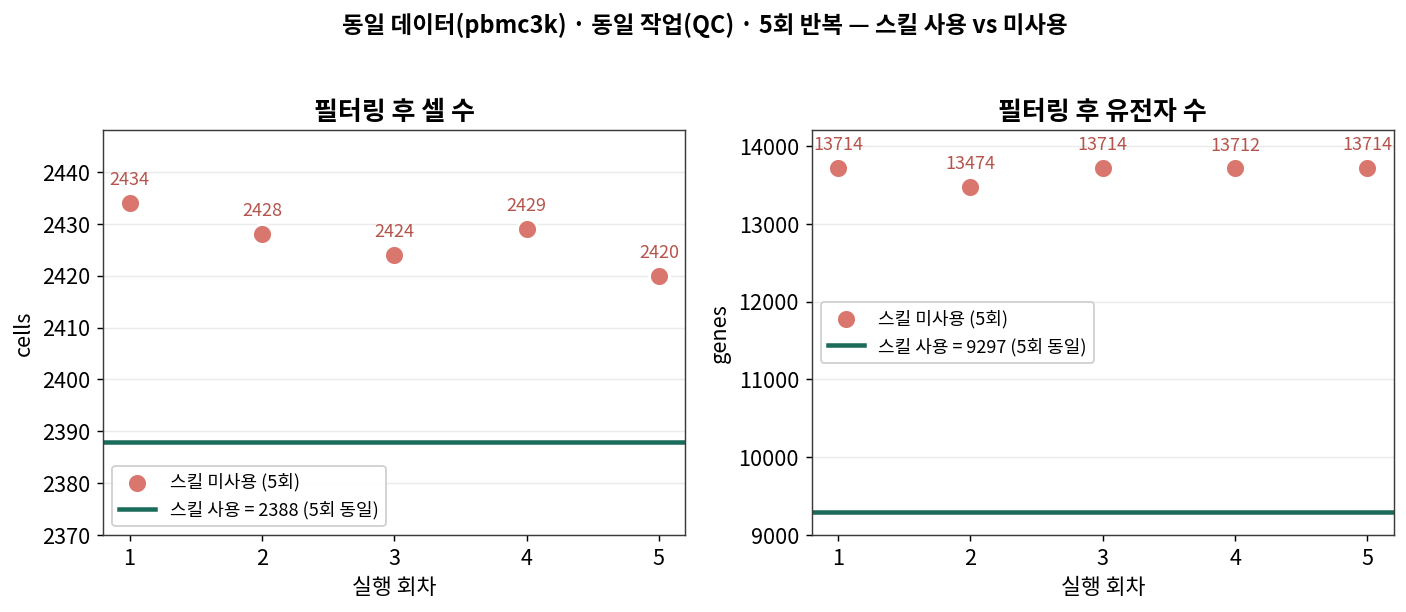
스킬 사용: 완벽하게 동일
2회 실행한 결과물(filtered.h5ad)을 비교하니, 셀 바코드·유전자·발현 행렬(X)이 전부 byte 단위로 일치했습니다.
SKILL run A == run B : obs_names ✓ var_names ✓ X matrix ✓
결과: 2,388 cells × 9,297 genes (매번 동일)
당연합니다. 고정된 스크립트니까요. 하지만 이 “당연함”이 바로 핵심입니다 — 스킬의 본질은 ‘검증된 방법론을 하나로 고정해 두는 것’이거든요.
스킬 미사용: 5번 다 다른 결과
| 실행 | 필터 후 셀 | 필터 후 유전자 | MT 컷오프 | 추가 변형 |
|---|---|---|---|---|
| run 1 | 2,434 | 13,714 | 3 MAD + >20% | — |
| run 2 | 2,428 | 13,474 | 3 MAD + >20% | zero-gene 미제거 |
| run 3 | 2,424 | 13,714 | 3 MAD + >8% | + top20 metric |
| run 4 | 2,429 | 13,712 | 3 MAD + >20% | + min_genes=200 |
| run 5 | 2,420 | 13,714 | 3 MAD + >8% | + top20 metric |
- 필터 후 셀 수가 5번 다 달랐습니다 (2,420 ~ 2,434, std ≈ 5.3).
- 유전자 수도 3종류로 갈렸고요 (std ≈ 107).
- 더 결정적으로, 단순히 “셀 개수“만 다른 게 아니라 남긴 셀이 누구냐(셀 집합의 정체성) 도 달랐습니다. 5개 실행을 둘씩 비교한 Jaccard 유사도가 평균 0.9965 — 평균적으로 두 실행이 남긴 셀의 약 0.35%가 서로 다른 셀이었습니다.
작아 보이죠? 하지만 “같은 데이터, 같은 작업, 같은 모델”인데 단 한 번도 똑같은 결과가 안 나왔다는 게 포인트입니다. 논문에 “QC 후 2,42X개 셀”이라고 적었는데 다시 돌리면 숫자가 바뀐다면, 그건 재현성 문제죠.
결과 2 — 정확성: 의외로 미묘했다
여기서 솔직하게 짚어야 할 게 있습니다. 스킬 미사용 에이전트들이 생각보다 일을 잘했습니다.
- 5개 에이전트 전부 고정 임계값(예: “MT > 5% 컷”)이 아니라 MAD 기반 outlier 검출을 선택했습니다. 이건 scverse가 권장하는 현대적 best practice예요.
- 5개 전부 여러 지표를 결합(joint) 해서 필터링했고, MT 유전자도
MT-prefix로 정확히 13개를 찾아냈습니다. - 5개 전부 시각화도 만들었고요.
즉, 요즘 모델은 스킬 없이도 “교과서적인 방법론”을 알고 있습니다. 큰 줄기(MAD 쓰자)는 알아서 수렴했어요. 차이가 난 건 세부 knob이었습니다:
- MT 하드 컷을 8%로 둘지 20%로 둘지
pct_counts_in_top_20_genes같은 추가 지표를 넣을지min_genes=200같은 사전 필터를 끼울지- 유전자 필터 기준을
min_cells=3으로 둘지
이 knob들이 흔들리면서 결과가 매번 달라진 겁니다. “방법론은 같은데 세부 설정값이 제각각” 이었던 거죠.
그럼 스킬은 왜 숫자가 더 다를까?
눈치채셨겠지만 스킬 결과(2,388 cells / 9,297 genes)는 미사용 평균(~2,427 / ~13,714)과 꽤 다릅니다. 이유는 스킬이 선택한 고정 파라미터 때문입니다:
- 유전자 필터가 더 공격적:
min_cells=20(에이전트들은 대부분min_cells=3) → 9,297개만 남김 - MT 하드 컷 8% + counts MAD를 선형 스케일로 계산 (median 2,197 / MAD 499 기준)
흥미로운 반전 하나. 에이전트들은 counts/genes에 log1p 변환 후 MAD를 적용했는데(오른쪽으로 치우친 분포엔 이게 더 통계적으로 타당합니다), 스킬은 raw 스케일에서 MAD를 계산했습니다. 이 디테일만 보면 에이전트 쪽이 약간 더 정교했다고도 볼 수 있어요.
그러니 “스킬 = 무조건 더 정확”은 과장입니다. 더 정확한 표현은 이렇습니다:
스킬은 근거 있는 하나의 답을 항상 똑같이 내놓는다. 에이전트는 여러 개의 (대체로 합리적인) 답을 매번 다르게 내놓는다.
그래서 정작 중요한 건 — 결과를 판단하는 사람
이 실험에서 제가 가장 크게 느낀 건 따로 있습니다. 스킬을 쓰든 안 쓰든, 결국 “이 결과가 합리적인가, 정확한가”를 판단하는 건 사람의 몫이라는 점입니다.
생각해보세요. 에이전트는 raw 스케일이 아니라 log1p 변환 후 MAD를 적용해서 사실 더 정교했고, 스킬은 min_cells=20으로 더 공격적인 유전자 필터를 걸었습니다. 둘 중 무엇이 내 데이터·내 연구 맥락에 더 타당한지는 숫자만 봐선 알 수 없습니다. 뉴런이나 심근세포처럼 MT%가 원래 높은 조직이라면 8% 하드 컷이 멀쩡한 셀을 다 날려버릴 수도 있고요.
스킬은 “검증된 방법을 고정”해줄 뿐, 그 방법이 지금 이 데이터에 맞는지까지 보증해주진 않습니다. AI가 그럴듯한 코드를 짜고 그럴듯한 숫자를 뱉어도, “MT 컷 8%가 이 조직에 맞나?”, “2,388개라는 셀 수가 생물학적으로 말이 되나?”, “왜 이 셀들이 버려졌지?”를 되물을 수 있는 사람이 없으면 — 재현성이 100%여도 재현 가능하게 틀린 결과가 될 수 있습니다.
🔑 자동화가 좋아질수록 역설적으로 더 중요해지는 능력은 “AI가 내놓은 결과를 보고 합리적인지·정확한지 판단하는 눈“입니다. 스킬은 분산을 줄여 그 판단을 쉽게 만들어줄 뿐, 판단 자체를 대신해주진 않습니다.
그래서, 스킬은 언제 가치가 있나
이번 실험이 말해주는 스킬의 진짜 가치는 “정확성 +α”가 아니라 이쪽입니다:
- 재현성·표준화 — 같은 입력 → 항상 같은 출력. 팀이 여럿이어도, 6개월 뒤에 다시 돌려도 동일. 논문·파이프라인엔 이게 생명입니다.
- 분산 제거 — “어떤 knob을 쓸까”를 매번 LLM의 즉흥 판단에 맡기지 않습니다.
- 완결성·이력 추적 — 스킬은 필터링 전/후 + 임계값 오버레이 시각화 3종, 그리고 QC 주석이 붙은 원본(
*_with_qc.h5ad)까지 자동 저장합니다. “왜 이 셀을 버렸나”를 나중에 되짚을 수 있어요. - 성능이 낮은 모델에서의 안전망 — 이번엔 고성능 모델(Opus 서브에이전트)이라 미사용도 MAD를 잘 골랐지만, 더 작은 모델이라면 “MT > 5% 컷” 같은 구식·자의적 방법으로 빠질 위험이 큽니다. 스킬은 그 바닥을 받쳐줍니다.
한계
- N=5는 작은 표본입니다. 분산의 방향성은 분명하지만 정밀한 통계량은 아닙니다.
- QC라는 비교적 표준화된 작업만 봤습니다. 클러스터링·세포 타입 주석처럼 선택지가 폭발하는 단계라면 미사용의 분산은 훨씬 커질 가능성이 높습니다.
- 고성능 모델로 테스트했기에 미사용 조건이 유리했습니다. 모델 성능이 낮을수록 스킬의 격차는 벌어질 겁니다.
- 스킬의 고정 파라미터가 모든 데이터에 최적은 아닙니다. 조직 특성(예: 뉴런·심근세포의 높은 MT%)에 따라 조정이 필요할 수 있어요.
결론
- 재현성: 스킬이 확실히 이깁니다. 같은 데이터·같은 작업인데 미사용은 5번 다 다른 결과가 나왔고, 스킬은 byte 단위로 동일했습니다.
-
정확성: 고성능 모델이라면 미사용도 best practice(MAD)에 수렴합니다. 스킬이 “더 똑똑한 방법”을 주는 게 아니라, “하나의 검증된 방법을 고정” 해주는 겁니다. (오히려 log 변환 같은 디테일은 에이전트가 더 나았던 지점도 있었고요.)
- 하지만 무엇보다: 스킬도 에이전트도 “이 결과가 내 데이터·내 맥락에서 합리적인지”까지는 보증하지 못합니다. 그 판단은 끝까지 사람의 일입니다.
한 줄 요약:
혼자 빠르게 탐색할 거면 스킬 없이도 괜찮습니다. 하지만 논문·협업·재실행되는 파이프라인이라면, 스킬은 “재현성 보험”으로서 값을 합니다. 그리고 어느 쪽이든, 결과를 보고 합리적인지·정확한지 판단하는 사람이 빠지면 둘 다 무용지물입니다.
개떡같이 던져도 AI가 그럴듯하게 해주는 시대지만, “그럴듯함”과 “매번 똑같음”은 다른 문제더라고요. 그리고 둘 다 갖춰도 마지막 한 칸 — “그래서 이게 맞는 결과인가”를 판단하는 사람 — 은 여전히 비워둘 수 없었습니다. 🧬
부록 — 재현 방법
# 데이터 준비
python3 -c "import scanpy as sc; sc.datasets.pbmc3k().write('pbmc3k_raw.h5ad')"
# 스킬 사용 조건 (Claude Code의 single-cell-rna-qc 스킬)
python3 qc_analysis.py pbmc3k_raw.h5ad --output-dir run_skill
# 기본값: MAD counts=5, genes=5, MT=3, hard MT=8%, min_cells=20
# 스킬 미사용 조건: 동일 데이터에 "best practice로 QC 해줘"만 주고
# 독립 에이전트를 N회 돌린 뒤 filtered.h5ad의 셀/유전자 수 분산 비교
환경: scanpy 1.12, anndata 0.12, Claude Code (Opus 4.8). 모든 숫자는 실제 실행 결과입니다.

댓글남기기