
AlphaGenome API로 변이 효과 예측하기
AlphaGenome이란?
AlphaGenome은 Google DeepMind가 개발한 DNA regulatory code 해석 모델입니다. 최대 100만 bp 길이의 DNA 서열을 입력받아 유전자 발현, 스플라이싱, Chromatin 구조 등 11가지 modality를 단일 염기 해상도로 예측합니다.
쉽게 말하면, 특정 변이가 유전자에 어떤 영향을 미치는지 AI로 예측해주는 도구입니다.
Avsec et al. “Advancing regulatory variant effect prediction with AlphaGenome” Nature 649, 1206–1218 (2026). DOI: 10.1038/s41586-025-10014-0
예측 가능한 Output
AlphaGenome은 하나의 모델에서 11가지 output을 예측합니다:
| Output | 설명 |
|---|---|
| RNA_SEQ | 유전자 발현 |
| SPLICE_SITES | 스플라이스 사이트 분류 |
| SPLICE_SITE_USAGE | 상대적 스플라이스 사이트 사용량 |
| SPLICE_JUNCTIONS | 스플라이스 정션 예측 |
| DNASE | Chromatin 접근성 (DNase-seq) |
| ATAC | Chromatin 접근성 (ATAC-seq) |
| CAGE | 프로모터 활성 |
| CHIP_HISTONE | 히스톤 변형 |
| CHIP_TF | 전사인자 결합 |
| CONTACT_MAPS | 3D Chromatin contact map |
| PROCAP | 초기 전사 활성 (PRO-cap) |
API 설정
API Key 발급
- https://deepmind.google.com/science/alphagenome 접속
- Google 계정으로 로그인
- API key 발급 (무료, 비상업용)
설치
pip install -U alphagenome
Python 3.10 이상 필요 (3.11 권장).
클라이언트 초기화
from alphagenome.models import dna_client
model = dna_client.create('YOUR_API_KEY')
Colab에서는 Secrets에 ALPHA_GENOME_API_KEY를 저장한 뒤:
from alphagenome import colab_utils
model = dna_client.create(colab_utils.get_api_key())
변이 효과 예측 실습
기본 구조
AlphaGenome의 변이 예측은 reference 서열과 alternate 서열을 비교하는 방식입니다.
from alphagenome.data import genome
from alphagenome.models import dna_client
model = dna_client.create('YOUR_API_KEY')
# 변이 정의 (1-based, VCF 형식)
variant = genome.Variant(
chromosome='chr22',
position=36201698,
reference_bases='A',
alternate_bases='C',
)
# 분석 구간 설정 (변이를 중심으로 1MB)
interval = variant.reference_interval.resize(dna_client.SEQUENCE_LENGTH_1MB)
# 예측 실행
outputs = model.predict_variant(
interval=interval,
variant=variant,
ontology_terms=['UBERON:0001157'], # 조직 ontology
requested_outputs=[dna_client.OutputType.RNA_SEQ],
)
# 결과: reference vs alternate 비교
ref_output = outputs.reference.rna_seq # REF 서열 예측
alt_output = outputs.alternate.rna_seq # ALT 서열 예측
임상 변이 예시: BRCA2 스플라이싱 변이
유방암/난소암 관련 유전자 BRCA2의 스플라이싱 변이를 예측합니다:
from alphagenome.models import variant_scorers
# BRCA2 스플라이싱 변이
variant = genome.Variant(
chromosome='chr13',
position=32316462,
reference_bases='T',
alternate_bases='G',
)
# 스플라이싱 관련 scorer 3종
splicing_scorers = [
variant_scorers.RECOMMENDED_VARIANT_SCORERS['SPLICE_SITES'],
variant_scorers.RECOMMENDED_VARIANT_SCORERS['SPLICE_SITE_USAGE'],
variant_scorers.RECOMMENDED_VARIANT_SCORERS['SPLICE_JUNCTIONS'],
]
interval = variant.reference_interval.resize(dna_client.SEQUENCE_LENGTH_1MB)
# 스코어링 실행
scores = model.score_variant(
interval=interval,
variant=variant,
variant_scorers=splicing_scorers,
organism=dna_client.Organism.HOMO_SAPIENS,
)
스플라이싱 통합 점수가 1.0 이상이면 유의미한 스플라이싱 영향이 있다고 판단합니다.
실제 실행 결과 (BRCA2 chr13:32316462 T>G, 전체 modality):
| Output | Max Raw Score | Quantile | Top Gene |
|---|---|---|---|
| CHIP_HISTONE | 114024.0 | 0.9997 | - |
| RNA_SEQ | 1.26 | 0.9996 | FRY |
| CHIP_TF | 2992.0 | 0.998 | - |
| SPLICE_SITE_USAGE | 0.02 | 0.995 | BRCA2 |
| CAGE | 32.1 | 0.990 | - |
| ATAC | 138.4 | 0.988 | - |
| PROCAP | 72.4 | 0.988 | - |
| DNASE | 155.9 | 0.985 | - |
| SPLICE_JUNCTIONS | 0.07 | 0.980 | BRCA2 |
| CONTACT_MAPS | 0.002 | 0.933 | - |
| SPLICE_SITES | 0.01 | 0.687 | BRCA2 |
BRCA2 스플라이싱 변이는 히스톤 변형(quantile 0.9997), 유전자 발현(0.9996)에서 극단적 영향이 예측되며, SPLICE_SITE_USAGE(0.995)와 SPLICE_JUNCTIONS(0.980)에서도 BRCA2 유전자에 직접적인 스플라이싱 영향이 확인됩니다.
배치 변이 스코어링
여러 변이를 한 번에 11가지 modality로 스코어링할 수 있습니다:
from alphagenome.models import variant_scorers
# 권장 scorer 11종 전체 사용
all_scorers = variant_scorers.RECOMMENDED_VARIANT_SCORERS
variant = genome.Variant(
chromosome='chr3',
position=58394738,
reference_bases='A',
alternate_bases='T',
)
interval = variant.reference_interval.resize(dna_client.SEQUENCE_LENGTH_1MB)
# 전체 modality 스코어링
variant_scores = model.score_variant(
interval=interval,
variant=variant,
variant_scorers=list(all_scorers.values()),
organism=dna_client.Organism.HOMO_SAPIENS,
)
# DataFrame으로 정리
df = variant_scorers.tidy_scores([variant_scores])
반환되는 DataFrame 컬럼:
| 컬럼 | 설명 |
|---|---|
gene_name |
영향받는 유전자 |
output_type |
예측 modality (RNA_SEQ, DNASE 등) |
variant_scorer |
사용된 scorer 종류 |
raw_score |
원시 점수 |
quantile_score |
분위수 점수 (0~1) |
시각화
from alphagenome.visualization import plot_components
import matplotlib.pyplot as plt
plot_components.plot(
[
plot_components.OverlaidTracks(
tdata={
'REF': outputs.reference.rna_seq,
'ALT': outputs.alternate.rna_seq,
},
colors={'REF': 'dimgrey', 'ALT': 'red'},
),
],
interval=outputs.reference.rna_seq.interval.resize(2**15),
annotations=[plot_components.VariantAnnotation([variant], alpha=0.8)],
)
plt.show()
REF(회색)와 ALT(빨간색) 트랙을 겹쳐서 변이가 유전자 발현에 미치는 영향을 시각적으로 확인할 수 있습니다.
아래는 실제 AlphaGenome API로 생성한 시각화 결과입니다.
BRAF V600E — RNA-seq REF vs ALT
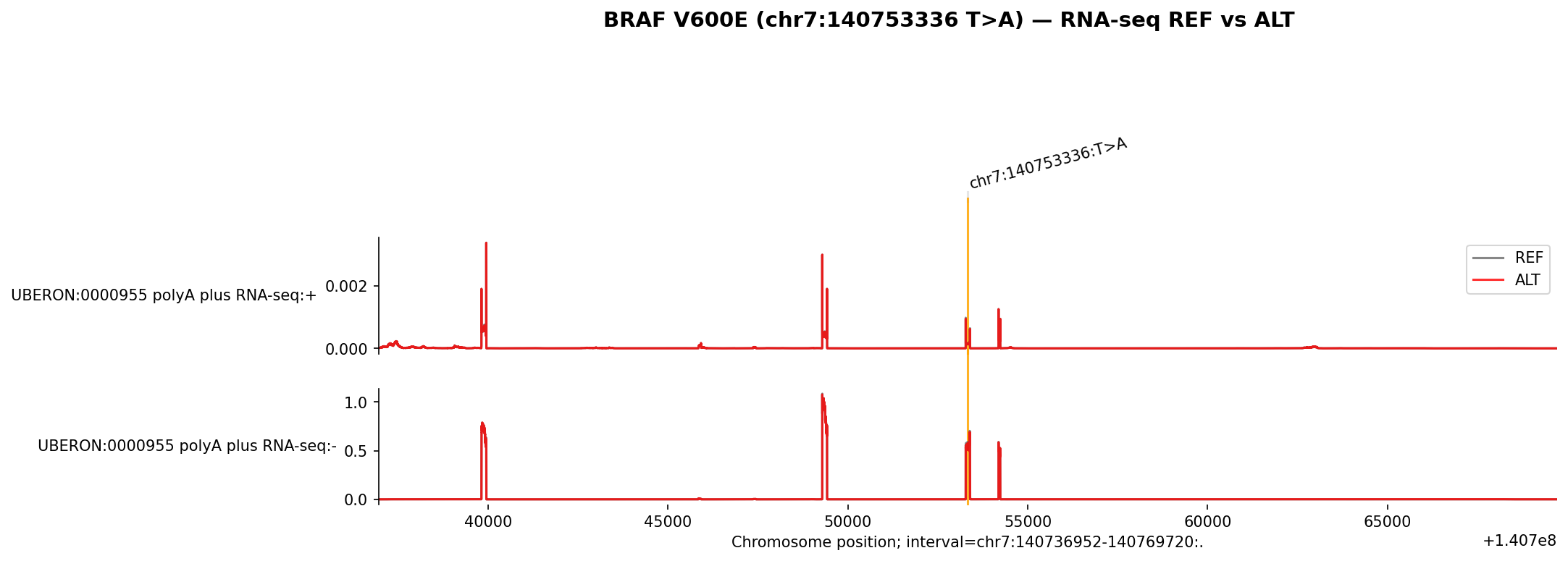
< BRAF V600E (chr7:140753336 T>A) — 변이 위치(노란색) 주변 발현 변화 >
BRCA2 스플라이싱 변이 — RNA-seq REF vs ALT
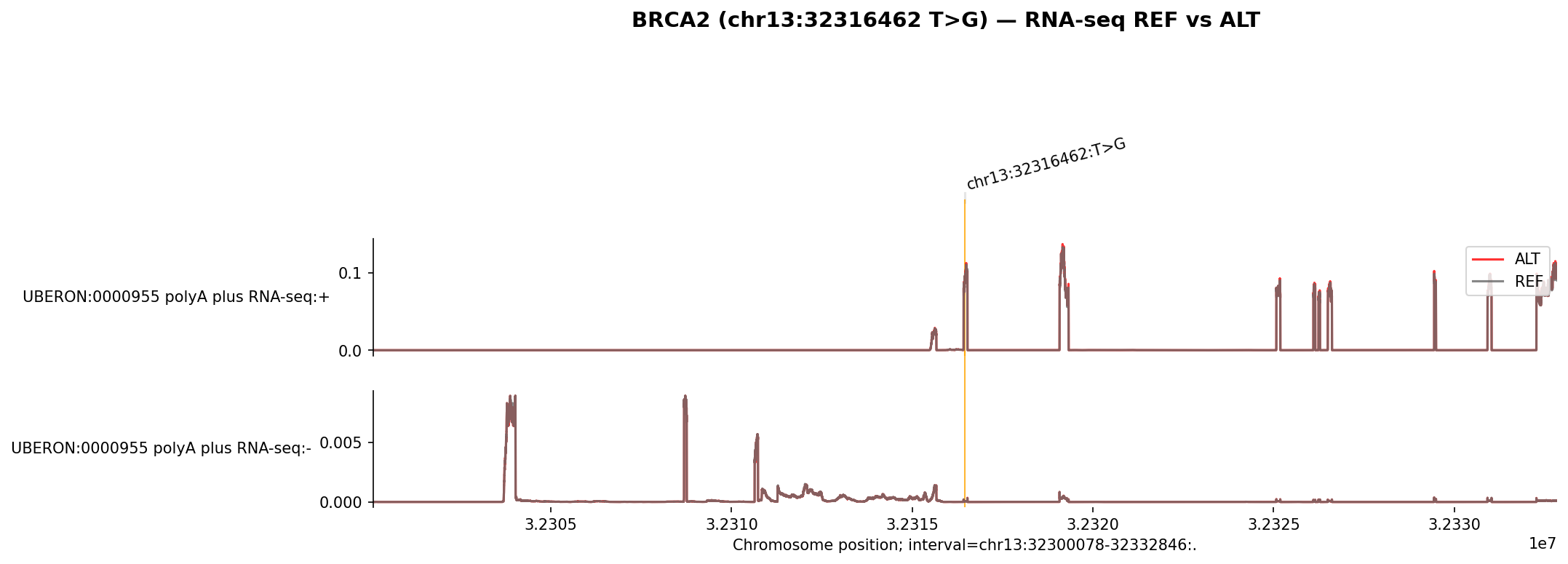
< BRCA2 (chr13:32316462 T>G) — 변이 위치 주변 발현 트랙 비교 >
지원 범위와 제한사항
| 항목 | 내용 |
|---|---|
| 지원 종 | Human (hg38), Mouse (mm10) |
| 서열 길이 | 16KB, 100KB, 500KB, 1MB (4가지) |
| 변이 유형 | SNV, insertion, deletion |
| 비용 | 무료 (비상업용) |
| 상업용 | 별도 신청 필요 (신청 페이지) |
| Rate limit | 수천 건 수준 적합, 100만 건 이상은 부적합 |
공식 리소스
- Colab 노트북 8개 제공 (GitHub):
quick_start.ipynb— 첫 예측batch_variant_scoring.ipynb— 배치 스코어링splicing_variant_scoring.ipynb— 스플라이싱 분석 (BRCA1, BRCA2, CFTR)example_analysis_workflow.ipynb— TAL1 종양유전자 분석visualization_modality_tour.ipynb— 11가지 modality 시각화
- YouTube: AlphaGenome 101
- 커뮤니티 포럼: https://www.alphagenomecommunity.com
한 걸음 더: MCP Tool로 만들기
여기까지는 Python 코드를 직접 실행하는 방식입니다. 하지만 이걸 MCP Tool로 만들면, LLM에게 자연어로 변이를 던지는 것만으로 AlphaGenome 결과를 받을 수 있습니다.
"BRCA2 chr13:32316462 T>G 변이의 스플라이싱 영향을 분석해줘"
→ LLM이 AlphaGenome MCP Tool을 호출
→ 스플라이싱 점수 + 영향받는 유전자 + 해석까지 자동 반환
MCP Server 만들기
MCP Server는 JSON-RPC 함수를 정의하면 됩니다. Python으로 간단한 AlphaGenome MCP Server를 만들어보겠습니다.
# alphagenome_mcp.py
from mcp.server.fastmcp import FastMCP
from alphagenome.data import genome
from alphagenome.models import dna_client, variant_scorers
import os
mcp = FastMCP("alphagenome")
model = dna_client.create(os.environ["ALPHAGENOME_API_KEY"])
@mcp.tool()
def score_variant(
chromosome: str,
position: int,
ref: str,
alt: str,
organism: str = "human",
) -> dict:
"""변이의 발현, 스플라이싱, Chromatin 영향을 예측합니다."""
variant = genome.Variant(
chromosome=chromosome,
position=position,
reference_bases=ref,
alternate_bases=alt,
)
org = (
dna_client.Organism.HOMO_SAPIENS
if organism == "human"
else dna_client.Organism.MUS_MUSCULUS
)
interval = variant.reference_interval.resize(
dna_client.SEQUENCE_LENGTH_1MB
)
all_scorers = variant_scorers.RECOMMENDED_VARIANT_SCORERS
scores = model.score_variant(
interval=interval,
variant=variant,
variant_scorers=list(all_scorers.values()),
organism=org,
)
df = variant_scorers.tidy_scores([scores])
# 주요 결과 요약
results = {}
for _, row in df.iterrows():
key = f"{row['output_type']}_{row['variant_scorer']}"
if key not in results or abs(row['raw_score']) > abs(results[key]['raw_score']):
results[key] = {
"gene": row.get("gene_name", "N/A"),
"output_type": row["output_type"],
"scorer": row["variant_scorer"],
"raw_score": round(float(row["raw_score"]), 4),
"quantile_score": round(float(row["quantile_score"]), 4),
}
return {
"variant": f"{chromosome}:{position} {ref}>{alt}",
"organism": organism,
"scores": list(results.values()),
}
if __name__ == "__main__":
mcp.run()
Claude Code에 등록
.mcp.json에 서버를 추가합니다:
{
"mcpServers": {
"alphagenome": {
"command": "python",
"args": ["alphagenome_mcp.py"],
"env": {
"ALPHAGENOME_API_KEY": "YOUR_API_KEY"
}
}
}
}
실제 사용 예시
등록 후 Claude에게 자연어로 질문하면 됩니다:
사용자: "BRAF V600E (chr7:140753336 T>A) 변이 효과를 분석해줘"
Claude: AlphaGenome score_variant 도구를 호출하겠습니다.
아래는 실제 AlphaGenome API 실행 결과입니다:
BRAF V600E (chr7:140753336 T>A)
| Output | Max Raw Score | Quantile | Top Gene | 해석 |
|---|---|---|---|---|
| CHIP_TF | 869.0 | 0.999 | - | 전사인자 결합에 극단적 영향 |
| RNA_SEQ | 3.95 | 0.998 | NDUFB2 | 유전자 발현 유의미한 변화 |
| CHIP_HISTONE | 7759.0 | 0.986 | - | 히스톤 변형 대규모 변화 |
| SPLICE_JUNCTIONS | 0.13 | 0.990 | BRAF | 스플라이스 정션 영향 |
| SPLICE_SITE_USAGE | 0.01 | 0.980 | BRAF | 스플라이스 사이트 사용 변화 |
| CAGE | 31.4 | 0.973 | - | 프로모터 활성 변화 |
| DNASE | 16.7 | 0.965 | - | Chromatin 접근성 변화 |
| PROCAP | 11.3 | 0.919 | - | 초기 전사 활성 변화 |
| ATAC | 12.4 | 0.682 | - | Chromatin 접근성 (ATAC) |
| SPLICE_SITES | 0.01 | 0.586 | BRAF | 스플라이스 사이트 분류 |
| CONTACT_MAPS | 0.001 | 0.494 | - | 3D 구조 영향 미미 |
BRAF V600E는 전사인자 결합(quantile 0.999), 유전자 발현(0.998), 히스톤 변형(0.986) 등 다수의 modality에서 극단적인 영향이 예측됩니다. 잘 알려진 발암 변이답게 광범위한 regulatory 영향을 보여줍니다.
Skill로 확장하기
MCP Tool이 “도구”라면, Skill은 “워크플로우”입니다. AlphaGenome Tool 위에 Skill을 얹으면 더 체계적인 분석이 가능합니다:
# AlphaGenome Variant Analysis Skill
1. 사용자가 변이를 입력하면:
2. score_variant 도구로 11가지 modality 스코어링 실행
3. quantile_score > 0.9인 항목을 "유의미한 영향"으로 분류
4. 영향받는 유전자와 modality를 요약
5. ClinVar, gnomAD 등 기존 데이터베이스와 비교 권장 사항 제시
6. 결과를 마크다운 테이블로 정리하여 반환
이렇게 하면 “BRCA1 변이 분석해줘”라는 한마디로 AlphaGenome 실행 → 결과 해석 → 임상 맥락 정리까지 자동으로 이루어집니다.
마무리
AlphaGenome은 변이 해석에 있어 기존 도구(CADD, SpliceAI 등)와 차별화되는 점이 있습니다. 하나의 모델에서 발현, 스플라이싱, Chromatin, 3D 구조까지 통합적으로 예측한다는 점입니다.
여기에 MCP Tool로 감싸면 코드를 몰라도 자연어로 변이 분석을 요청할 수 있고, Skill까지 추가하면 분석 워크플로우 자체를 자동화할 수 있습니다.
API → Tool → Skill. 이 세 단계가 “AI한테 개떡같이 던져도 찰떡같이 나오는” 구조의 핵심입니다.




댓글남기기