
Hail - (4) Hail Table
Hail Table
Hail에서 Table은 우리가 흔하게 알고 있는 SQL의 table, pandas 또는 R의 Dataframe 등과 매우 유사하다고 보시면 됩니다. row field(column)마다 type(string, int32, array 등)이 지정되어 있는 schema에 따라 데이터가 여러 행으로 구성되어 있습니다. 차이가 존재하다면 row fields 이외에 global fields가 존재한다는 것과 Spark의 데이터처럼 파티션으로 나뉘어져 있다는 것입니다. global fields는 Table에 대한 전체적인 메타 정보를 저장할 수 있다는 것이 전부입니다. 크게 어렵지 않죠?
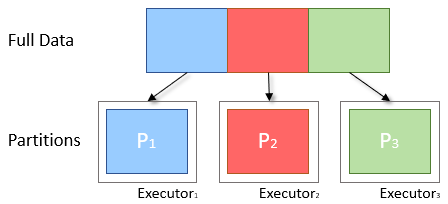
< Partition의 개념 >
파티션에 대해 간단하게 말하자면 병렬처리를 위해 데이터를 나누는 단위라고 할 수 있습니다. 1개의 파티션마다 1개의 작업(task)이 부여가 되고 1개의 코어가 이 작업을 처리합니다. 따라서 코어의 개수에 따라서 파티션의 개수를 설정하면 좋은데, 일반적으로 코어 수의 2~4배가 가장 좋다고 알려져 있습니다. 그러나 이것은 모든 상황에서 이상적인 값이라고 할 수는 없기 때문에 Spark 관련 경험과 지식을 습득하여 적절한 파티션의 수를 찾는 것이 좋을 것 같습니다.
이해를 돕기 위해서 gene expression file과 gene location file을 예제로 사용하려고 합니다. 그럼 Hail Table을 읽는 방법부터 시작해서 하나씩 알아보도록 할까요?
1) 읽기 (Read & Import)
우선 데이터를 Table 형태로 불러오는 것부터 시작하겠습니다.
이미 Hail Table로 저장이 되어 있다면 불러오는 것은 간단합니다. read_table 함수로 말이죠.
import hail as hl
ht = hl.read_table('path/to/hail_table.ht')
하지만 대부분의 데이터는 얻게 되는 경로는 텍스트 파일일 가능성이 높습니다. 따라서 처음에는 import_table 함수를 자주 쓰게 되죠. 유용한 parameter가 많기 때문에 자세히 보도록 하겠습니다.
hail.methods.import_table(paths, key=None, min_partitions=None, impute=False,
no_header=False, comment=(), delimiter='\t', missing='NA', types={},
quote=None, skip_blank_lines=False, force_bgz=False, filter=None,
find_replace=None, force=False, source_file_field=None)
key와 partition에 대해서는 조금 있다가 살펴보기로 하고 우선 impute에 대해서 설명하겠습니다. 기본값은 impute=False인데 모든 data type을 str으로 간주하여 불러들이게 됩니다. impute=True로 설정하게 되면 Hail이 알아서 field의 type을 유추하여 schema를 구성하게 됩니다. 가지고 있는 data type에 숫자가 많은 경우에는 유용한 parameter가 되겠죠.
그 다음으로 설정하면 좋은 parameter가 min_partitions입니다. min_partition이 기본값인 경우에는 Hail은 Table의 크기에 따라서 파티션의 개수를 정해줍니다. 보통 이 정해진 개수는 사용할 수 있는 코어의 수보다 낮은 경우가 많죠. 용량이 적다면 파티션 수가 적어도 소수의 코어로 그 Table만을 작업하는 것은 시간이 오래 걸리지 않습니다. 오히려 더욱 효율적일 수도 있죠. 하지만 용량이 어느정도 커지게 되면 가능한 모든 코어를 사용하는 것이 더 유용합니다. 따라서 데이터의 용량이 크다면 min_partition parameter를 이용하여 파티션을 코어의 수 이상으로 정하는 것을 추천드립니다.
parameter 중 주의해야 할 것은 바로 force_bgz와 force입니다. 텍스트 파일의 경우 용량이 크다면 압축을 하게 되는데, Bioinformatics에서 주로 사용하는 압축 포맷이 bgz입니다. 그런데 bgz가 조금 간사한 게 확장자를 gz로 둔갑시켜 헷갈릴 때가 많습니다. 파일의 확장자가 bgz이면 import할 때 이를 압축파일임을 자동으로 인식하여 불러들이지만, 확장자가 gz인 경우에는 이를 인식하지 못하거든요. 이때 force_bgz=True 설정을 통해 bgz 압축 파일임을 인식하도록 할 수 있습니다. 실제로 gz 압축 파일이면 어떻게 해야 할까요? force=True를 이용해서 불러들이면 되지만 1개의 core만을 사용하기 떄문에 상당히 비효율적입니다. 따라서 gz 압축 파일인 경우에는 압축을 gunzip이나 unpigz(multi thread를 사용하여 속도가 빠름)를 통해 압축을 먼저 풀어주고 읽는 것을 추천합니다.
그 외에 몇몇 parameter가 어떻게 사용되는지는 예를 들고 넘어가겠습니다. import하려는 파일의 header가 존재하지 않을 때, no_header=True로 설정하면 임의로 field name(f0, f1, … fN)을 정하고 텍스트 파일의 첫째 행부터 불러들입니다. 또한 만약 파일의 구분자가 ,인 경우에는 delimiter=','으로 설정해주면 되고, 파일에 NaN이 missing value로 표기되어 있다면 missing='NaN'을 사용하면 됩니다. 그 외의 parameter도 거의 pandas나 R과 비슷하게 설정되어 있다는 것을 알 수 있습니다.
예제로 gene expression file과 gene location file을 Hail Table로 import를 해보겠습니다.
## Import Tables
gene_expr = hl.import_table('gene_expr_join.tsv', impute=True)
gene_loc = hl.import_table('gene_loc_join.tsv', impute=True)
불러온 Hail Table의 내용을 확인하고 싶다면 show 함수를 사용해봅니다.
gene_expr.show()
gene_loc.show()
impute=True 옵션 덕분에 숫자형 데이터의 경우 float64 또는 int32 data type이 지정된 것을 확인할 수 있습니다.
2) Table 병합에 중요한 key
어떤 언어든 Table 형식의 데이터를 다룰 때 자주 사용하는 것이 바로 여러 Table을 병합(join)하여 Table 간의 정보를 연결하는 것입니다. 가령, gene expression 데이터를 가지고 있는데 gene에 대한 위치 정보를 얻기 위해 GENCODE와 같은 데이터베이스에서 파일을 구해 병합할 수 있을 것입니다. 이 때 사용되는 것이 바로 key입니다. key는 SQL의 Key와 같은 개념이라고 이해하시면 될 것 같습니다. key를 이용하면 병합 뿐만 아니라 semi_join, anti_join 등을 통해 다른 Table과 key가 겹치는 행만 얻거나 뺄 수 있습니다. 간단한 예제를 보겠습니다. 두 개의 Hail Table을 병합을 하는 과정입니다.
우선 key_by 함수를 통해 Table의 key를 지정해야 합니다. 위의 두 Table을 보면 gene_expr의 id field와 gene_loc의 geneid field가 같은 속성을 지니고 있습니다. 공유하고 있는 field를 key로 지정하면 두 Table을 잇는 작업을 할 수 있게 되지요.
다음은 gene_expr Table에 대해서 key를 지정하는 코드입니다. describe 함수를 통해서 key가 잘 설정이 되었는지 확인할 수 있습니다. key_by에 대한 인수로 'id'처럼 field name을 바로 입력해도 되고, 또는 gene_expr.id처럼 attribute 형태로 입력해도 됩니다.
gene_expr = gene_expr.key_by('id')
gene_expr.describe()
----------------------------------------
Global fields:
None
----------------------------------------
Row fields:
'id': str
'Sam_01': float64
'Sam_02': float64
'Sam_03': float64
'Sam_04': float64
'Sam_05': float64
----------------------------------------
Key: ['id']
----------------------------------------
gene_loc Table 역시 key를 설정해줍시다.
gene_loc = gene_loc.key_by(gene_loc.geneid)
gene_loc.describe()
----------------------------------------
Global fields:
None
----------------------------------------
Row fields:
'geneid': str
'chr': str
'start': int32
'end': int32
----------------------------------------
Key: ['geneid']
----------------------------------------
이제 join 함수를 통해서 Table을 병합해보면 다음과 같습니다.
## Join the Tables (Inner Join)
join_table = gene_expr.join(gene_loc)
join_table.describe()
----------------------------------------
Global fields:
None
----------------------------------------
Row fields:
'id': str
'Sam_01': float64
'Sam_02': float64
'Sam_03': float64
'Sam_04': float64
'Sam_05': float64
'chr': str
'start': int32
'end': int32
----------------------------------------
Key: ['id']
----------------------------------------
다른 Table로부터 field 일부만 가져오고 싶으면 field를 새로 생성하는 annotate 함수를 사용합니다. 이때, Hail을 처음 배우는 분들에게는 헷갈리는 지점이 바로 다음 예에서 보이는 gene_loc[gene_expr.id]입니다. 이는 gene_loc에 대해서 gene_expr의 key인 id로 mapping된 StructExpression입니다. struct에 대해서는 조금 생소하실지 모르지만 간단하게 말씀드리면 여러 field를 포함하는 구조입니다. 예를 들어 struct = hl.struct(a=5, b='Foo')라고 정의가 되어 있다면 struct field 안에 a, b field를 포함하게 되는 것입니다. 그리고 struct.a 또는 struct['a']로 struct 안의 field를 호출할 수 있습니다. 따라서 gene_loc의 field 중 chr field를 가져오고 싶다면 다음과 같이 코드를 작성하면 됩니다. 이 코드에서 재밌는 사실은 gene_expr의 경우 따로 key를 지정하지 않아도 다른 Table의 key만 지정되어 있다면 field를 옮겨올 수 있다는 점입니다. 굳이 두 Table의 key를 맞춰주지 않아도 되기 때문에 편리한 방법입니다. 참고로, key_by 함수에 인수가 들어가지 않으면 key가 존재하지 않는 Table이 만들어집니다.
## Annotate a Field (chr) from gene_loc Table
gene_expr = gene_expr.key_by()
annot_table = gene_expr.annotate(chr = gene_loc[gene_expr.id].chr)
annot_table.describe()
----------------------------------------
Global fields:
None
----------------------------------------
Row fields:
'id': str
'Sam_01': float64
'Sam_02': float64
'Sam_03': float64
'Sam_04': float64
'Sam_05': float64
'chr': str
----------------------------------------
Key: ['id']
----------------------------------------
key와 관련된 함수 중 semi_join 역시 자주 사용하는 함수입니다. 만약 gene expression 데이터 중 위치 정보를 가지고 있는 gene에 대해서만 발현량을 보고 싶다면 다음과 같이 코드를 작성해볼 수 있겠네요.
우선, sample이라는 함수로 gene_loc Table을 Downsampling하여 몇개의 gene만 선택합니다.
## Downsample gene_loc Table
gene_loc = gene_loc.key_by('id')
filtered_gene_loc = gene_loc.sample(0.5)
filtered_gene_loc.show()
그리고 semi_join 함수를 통해 gene_expr Table을 Downsampling된 filtered_gene_loc에 존재하는 gene에 대해서만 subset할 수 있습니다.
## Filter gene_expr Table through key in filtered_gene_loc Table
gene_expr = gene_expr.key_by(gene_expr.id)
filtered_gene_expr = gene_expr.semi_join(filtered_gene_loc)
filtered_gene_expr.show()
위의 annotate 함수의 예와 마찬가지로 subset하려는 Table의 key를 다른 Table과 맞추지 않고도 subset하는 방법도 있습니다. 바로 filter라는 함수를 이용하는 건데요, 이 함수의 인수로 들어가는 값이 True인 행은 포함하고 False이면 제외하는 방식입니다. is_defined 함수는 인수로 들어가는 Expression이 결측값이 아닌 정의된 값이라면 True, 그렇지 않다면 False를 return합니다. 결국에는 filtered_gene_loc에 존재하는 gene만 정의되어 있기 때문에 filtered_gene_expr Table은 gene에 대한 expression 값을 갖게 되겠네요.
gene_expr = gene_expr.key_by()
filtered_gene_expr = gene_expr.filter(hl.is_defined(filtered_gene_loc[gene_expr.id]))
filtered_gene_expr.show()
3) 쓰기 (Write & Export)
Table을 읽는 것을 알게 되었으니 쓰는 것도 배워 볼까요? 우선 Hail Table 형태로 저장하는 방법은 write 함수를 이용하는 것입니다. 주의할 점이 있다면 Hail Table을 읽을 때 사용했던 read_table의 경로와 똑같은 경로로 저장을 할 수 없다는 것입니다. 이는 예측하건대 데이터를 읽으면 모든 데이터를 memory에 올리는 R이나 pandas 같은 프로그램과 달리 데이터를 다 memory에 올리는 것이 아니라 필요할 때마다 데이터에 접근하는 Spark의 특성 때문이라고 생각하고 있습니다. 또한 쓰려고 하는 경로에 이미 Hail Table이 존재한다면 error가 발생하는데 overwrite=True를 설정하면 덮어쓰기를 할 수 있습니다.
ht = ht.write('path/to/hail_table.ht', overwrite=True)
하지만 다른 사람들에게 분석의 결과물을 공유할 때는 텍스트 파일로 변환하는 경우가 많겠죠? Table을 텍스트 파일로 저장하고 싶다면 쓸 수 있는 함수가 export입니다. export는 write와 마찬가지로 import_table에 사용한 경로에 다시 저장을 하는 것이 불가능합니다. 다른 점이 있다면 write와 다르게 저장하려는 경로에 텍스트 파일이 존재한다면 기본적으로 들여쓰기를 하게 됩니다. 또한 delimiter='\t'가 기본 설정이기 때문에 csv 파일로 저장하려면 delimiter=',' 설정을 하면 되겠네요. 마지막으로, 텍스트 파일이 너무 크다면 저장하려는 파일 경로 끝에 .bgz 확장자를 붙이면 자동적으로 bgz 압축 파일로 만들어줍니다. 그리고 .gz 확장자를 뒤에 붙이면 gz 압축 파일이 되겠지요.
gene_expr와 gene_loc를 병합한 join_table을 tsv 파일로 저장하는 코드는 다음과 같습니다.
# Export the Table
join_table.export('join_table.tsv')
참고로 export 함수의 경우 Table 전체에서만 쓸 수 있는 것이 아니라 field 하나에 대해서만 저장할 때도 유용하게 사용할 수 있습니다. 예를 들어 gene_loc의 geneid field에 대해서만 텍스트 파일로 저장하고 싶다면 다음과 같이 코드를 작성할 수 있겠습니다.
gene_loc.geneid.export('geneid.tsv')
쓰다보니 내용이 무척 많아졌네요... 다뤘던 예제에 대한 notebook 파일도 첨부하겠습니다.
다음 시간에는 Matrix Table을 예제를 통해 다뤄보도록 하겠습니다.



댓글남기기